How can we be confident that the tree is robust with respect to the inputs? Is the tree reflecting the global phylogenetic signal or is it under the heavy influence of one character?
The way the analysis has performed is already a good sign on the robustness of the result: are there many equally parsimonious trees? Are the consensus trees well resolved with many nodes above 70%? Did the parsimony search converged rapidly?
But there are more objective tools. One kind tests the tree structure itself, another kind measures the compatibility of the characters with the tree structure.
Test the structure
The two tests most widely used for the tree structure are the bootstrap and the decay (or Bremer) computations.
The bootstrap (HOLMES 2003) is a random draw of characters with replacement. Hence you generate a matrix with the same size by selecting characters (columns) randomly. You may end up with some characters occurring more than once and some characters missing. You do the same analysis with the same software and options, and repeat the process. At the end, you compute a consensus tree (majority-rule) and give to each node the percentage of occurrence. Above 95% you have a very robust node, whereas under 50% the node is barely significant. The bootstrap test generally considers 1000 such random draws with replacement, and as a result can take a lot of computing time. Note that this technique is not very relevant when the number of characters is low.
The other useful test, the decay or Bremer degeneracy index, is defined as the number of supplementary steps necessary to have the node disappear. In other words, it looks at the trees that are slightly more complicated than the most parsimonious one, and compute the difference in total number of steps between the two trees when the node disappears. This test has the advantage of relaxing somewhat the parsimony criterion and keeps an eye on trees that are not so much more complicated than the most parsimonious one and not so much different.
Test the characters on the tree
The other robustness tests evaluate the compatibility of characters with the tree. They try to quantify how much each character supports the tree structure, or how much it is responsible for it. This indexes can be computed for each character or for all.
Let’s first define the following quantities for each character:
: number of steps for the tree to test
: minimal number of steps that it is possible to get with the dataset. It is computed by disregarding the homoplasies (reversals, convergences and parallel evolutions)
: maximal number of steps that it is possible to get with the dataset. It corresponds to the minimum number of steps of a fully non-resolved tree (star tree) when there is no phylogenetic signal.
These quantities can then be estimated globally by adding the values for each character. To evaluate the fitness of the characters on the tree, the following indexes are computed:
- Consistency Index:
- Homoplasy Index:
- Retention Index:
- Rescaled Consistency Index:

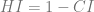


The Retention Index RI measures the ratio between the real number of synapomorphies to the apparent one. The Rescaled Consistency Index RCI is a better indicator than the simple Consistency Index CI since it is more reliable by eliminating autopomorphies.
Ideally, CI, RI and RCI should be close to 1 and HI close to 0, with 
However, these indexes depend a lot on the number of taxa (for instance, CI decreases when this number increases). As a consequence, they have no value in absolute and are useful to compare several trees for the same dataset.
For each character, they give a valuable indication on the fitness of the character in the hypothetical phylogeny given by the tree. These indexes can thus compare the characters one to each other.
Anyhow, I favor the projection of the character values on the tree since it gives a better physical understanding of the fitness of the characters and the relevance of the proposed phylogeny. But I admit that it is more subjective…
——————————————————–
HOLMES, S., 2003. Bootstrapping phylogenetic trees : Theory and methods. Statistical Science 18, 241–255.

